The problem
Alzheimer’s disease (AD) is a leading cause of death in the UK and along with other dementias, is responsible for almost 11% of all deaths in England.1 More than 209,000 new cases are diagnosed each year across the UK.2 It remains a leading cause of death and disability worldwide3, affecting nearly 50 million people. Every three seconds someone develops dementia, equating to 9.9 million new cases annually worldwide.2
Billions have been spent on AD research to date. Since 2008, the US National Institutes of Health (NIH) alone has spent over $32 billion on research grants for AD and other dementias, with projections for 2022 and beyond expected to be much higher. 4 Between 2010 and 2020, UK government investment in dementia research has increased from £28.2m to £75.7m per year.5 Since 1998, the charity Alzheimer’s Research UK has provided £171 million to research projects.6
Despite the scale of funding, finding new treatments for AD has proved virtually impossible and has been described as a ‘graveyard for expensive drug tests.7 Hundreds of drugs have been developed but have an appalling failure rate during clinical (human) trials of over 99%, representing a dismal return on global investment into AD research.8 Failures are due to either a lack of proof that the drug works (efficacy) or safety related side effects (toxicity).9 There are a few drugs available to AD patients, however they too may cause side effects10 and are offered to try to manage day to day symptoms at best. There is no drug available which can slow or stop the worsening of AD.
Controversy and the AD crisis
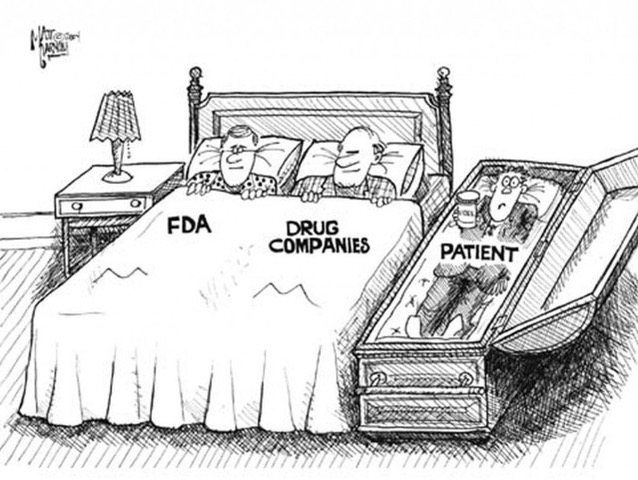
No new treatments were approved for AD between 2003 and 2021, until the controversial approval of Aduhelm (aducanumab) by the US Food and Drug Administration (FDA) last June.11 The approval was highly criticised by the FDA’s own advisory committee and some staff resigned, allegedly in protest over concerns about Aduhelm’s safety and efficacy and that its approval had been ‘fast tracked’ without appropriate evidence of its benefit to patients.12
Aduhelm’s mechanism of action is to target ‘beta amyloid’, a type of protein which gathers to form sticky clumps or ‘plaques’ between brain cells and is considered to correspond to the decrease in brain function and cognitive decline seen in AD. Most research has focused on blocking this protein to stop plaques accumulating. Another is a protein called ‘tau’ present in the nerve cells of the brain (neurons). In healthy brains, tau proteins are long and straight, to enable transport of nutrients along and between neurons. In patients with AD, these tau proteins are seen to collapse and become tangled, so nutrient transport breaks down and the neurons die. However, research into these proteins and the link to AD is not straightforward, as plaques and/or tangles are known to form long before the onset of AD symptoms.13 Drug failure rates may be compounded by the fact that by the time AD patients enter trials, brain cell damage may be too advanced, leading to some research focus shifting to earlier in the disease process, for example new vaccine trials.14 Furthermore, some patients diagnosed with AD are found to have few or no plaques in the brain15 and plaques can be present in people who never develop AD.16
Following Aduhelm’s approval, AD research has again attracted controversy very recently, with media coverage raising concerns that data from a high-profile study carried out in mice and rats into a particular protein in the brain called ‘amyloid beta *56’ may have been manipulated or exaggerated. Concerns are intensified as the 2006 study was highly influential when published in Nature and has since been cited by well over 2,000 further studies, resulting in multi-million-dollar grants being issued for research on the same theory. 17 For example, as explained in a recent Science article which reported the story, in the last financial year alone the NIH has allocated $1.6 billion (approximately half of its AD funding) to ‘amyloid’ related projects and annual NIH support for similar studies increased from near zero to $287 million by 2021.18 The amyloid theory has dominated AD research to the general exclusion of most other areas of investigation13 and may have potentially misled the field for almost two decades,19 compounded by evidence of a long list of failed treatments.
Why is the failure rate so high?
Prior to being tested in human (clinical) trials, all potential new drugs are assessed in preclinical (also sometimes called ‘nonclinical’) tests for safety and efficacy, the vast majority of which are performed in animals. It’s important to note that animals are not just used at the preclinical stage, but also throughout the entire earlier process of AD research to study human disease mechanisms, identify new therapeutic targets, and develop new drugs before testing them.20 Animal tests are performed either before, or in parallel to several stages of clinical trials in humans. However, nearly all potential new AD treatments have failed somewhere along the clinical trial process, leading to the attrition rate described above. For example, mice often respond to AD drugs, leading to overestimation of performance and subsequent rejection during clinical trials, when humans do not produce the same response. Furthermore, some experimental drugs have even been considered to worsen cognitive decline in patients.21
Animals do not naturally develop AD (and many other diseases) in the same way as humans. This is well known and has led to a dramatic increase in the use of genetically modified (GM) animals to try to artificially create disease symptoms and ‘humanise’ them. However, while GM animals such as mice can be engineered, for example to develop the ‘tau’ protein tangles seen in the brain cells of AD patients, their underlying biology means that results have not translated to patients in the clinic, leading many researchers to conclude that better science is needed. 22
Sadly, research (and those who regulate and approve it) remains entrenched in the use of animal models, despite the known failures. The NIH National Institute of Aging recently said, in reference to AD: “while we have been able to cure the disease in mouse models of the disease, we have not been able to translate these advances to humans”. But instead of this prompting a shift towards more human relevant strategies, the institute is looking for ‘alternative animal models’ such as ‘better mouse models’ and commenting, “the marmoset shows promise”, while looking to recruit “early career investigators interested in expanding the range of animal model paradigms”.23 It’s important to clarify that many species of animals including mice, rats, dogs and monkeys have been used for decades and an animal model termed as ‘new’ usually means trying a different mutation or strain. Old ways and out of date methods – even when they don’t deliver – are clearly hard to escape from, especially when high figure grants are at stake and research infrastructure is firmly embedded in global business supply chains involved in the ongoing breeding, supply and transport of animals. This promises yet another eventual dead end and is astonishing, given how much of the NIH yearly spend is on AD. Animals cannot adequately replicate the disease in humans and so in turn are unable to predict human responses to drugs, due to extensive species differences from macro to micro (molecular) levels. Simply put, they are not human-relevant.
This presents a two-fold problem; firstly, decades of continued reliance on animals keeps leading to failure in human trials and secondly, patients are potentially missing out on many effective treatments that have been rejected, based on a lack of desired response during animal tests. Sadly, this is not a problem confined to AD. The general failure rate for new drugs is between 86-95%. Although several thousand diseases affect humans, only about 500 have any approved treatments.24
Human relevant science is the solution
There is a wide range of human-relevant scientific methods which can be employed in Alzheimer’s Disease research. For example, the pioneering in vitro ‘organ on a chip’ system uses human cells and tissues in microfluidic devices. These mimic a variety of human organs to test new compounds before entering clinical trials. With specific application to AD, this technology has been developed using neurons derived from human stem cells, to provide a model which more closely mimics the development of human AD – and therefore response to treatments – than animals.25
In response to the urgent need to address the lack of effective treatments for AD and the failure of conventionally used (animal) models to date, the Joint Research Centre (JRC) of the European Commission recently published a comprehensive database of 567 biochemical and computational research models (the JRC has also developed similar databases for hundreds of new methods in other disease areas). The publication was a result of the centre’s ongoing work to ‘provide an inventory and scientific evaluation of innovative (human-based) non-animal models/approaches currently in use for basic and applied research in the field of neurodegenerative diseases, more specifically Alzheimer’s and Parkinson’s disease’, and to ‘contribute to the increased adoption and acceptance of alternative methods in neurodegeneration research and related fields. Just a few examples include 3D organoid, ‘brain on a chip’ or in silico (computational screening) methods to investigate protein aggregation or neuroinflammation.26
There is also scope to vastly improve the use of libraries of existing observational, clinical and real-world evidence (RWE) data on AD, given the many hundreds of trials and studies over decades. Retrospective analysis and systematic review of this data can provide opportunities for better diagnosis, treatment and prevention strategies27 as well as human systems biology-based approaches. As proposed in the Journal of Alzheimer’s Disease28, a massive international human genome sequencing effort could help to elucidate the fundamental nature of AD and resolve whether the amyloid hypothesis or other competing hypotheses, such as the innate immunity hypothesis, is correct.
As described earlier, the chronic pathological processes leading to brain cell breakdown may mean that AD is too advanced by the time patients are diagnosed, which has been speculated as a contributing factor to drug failures. For example, some human-relevant models recognise this and are built to address the earlier stages of mild cognitive impairment (MCI) which can precede AD in patients.29
There are other factors in brain physiology and function which may contribute to AD. For example, improvements in cardiovascular health are known to decrease risk of dementia 30 and lifestyle factors such as maintaining good physical and mental activity and a healthy diet remain critical.31 In fact, the US National Plan to address AD and related dementias was updated in 2021 to include actions for promoting healthy aging by reducing risk factors, including physical inactivity, hypertension, smoking or excessive alcohol drinking, unhealthy diet, diabetes, infectious diseases, exposure to toxins, physical brain trauma, depression, and low cognitive/social/educational attainments, among many other damaging factors and conditions associated with aging.32
These are just a few examples of the types of human-relevant approach that can be taken forward in AD research. Many are working in this area, but maintaining funding to prove that this is a better scientific way forward remains challenging. The need for government action, laws and regulatory attitudes to catch up is long overdue.
The opportunities to transform AD research are not limited to just one approach or data source. Instead, all available human-relevant methods can and must be used in combination as ‘new approach methods’ (NAMs) to provide real world solutions, and to end over-reliance on animal models which are failing to deliver the treatments so urgently needed by AD patients. This closing comment by a senior editor of the Journal of Alzheimer’s Disease is aimed at the amyloid hypothesis – but it could equally be applied to the animal model paradigm:
“It is unacceptable, in my judgment, when medical researchers (for whatever reasons) steadfastly hold onto a hypothesis that does not help sick patients in any manner despite being paid to do it. Rationalizing such behavior blocks medical progress resulting in dire consequences for the patients’ clinical outlook. Equally disturbing is the callous effect such conduct has on devaluing the scientific spirit and the search for truth.” 33
References
- Monthly mortality analysis, England and Wales – Office for National Statistics (ons.gov.uk)
- Incidence in the UK and globally – Dementia Statistics Hub.
- 2022 Alzheimer’s disease facts and figures – PubMed (nih.gov)
- RePORT (nih.gov)
- Research funding – Dementia Statistics Hub
- About our research – Alzheimer’s Research UK (alzheimersresearchuk.org)
- Failure Upon Failure For Alzheimer’s Drugs | Inside Science
- World Alzheimer’s Day: New approach methodologies are urgently needed – Safer Medicines
- Alzheimer’s disease drug-development pipeline: few candidates, frequent failures | Alzheimer’s Research & Therapy | Full Text (biomedcentral.com).
- Effects of Alzheimer’s disease drugs | Alzheimer’s Society (alzheimers.org.uk)
- New drug for Alzheimer’s – Safer Medicines
- Three FDA advisers quit over agency approval of aduhelm (medicalxpress.com)
- The amyloid hypothesis on trial (nature.com)
- The inside story of the search for an Alzheimer’s vaccine (telegraph.co.uk)
- Mild to moderate Alzheimer dementia with insufficient neuropathological changes – PubMed (nih.gov)
- Plaques, Tangles in Brain Don’t Always Lead to Alzheimer’s (medicinenet.com)
- A specific amyloid-β protein assembly in the brain impairs memory | Nature
- https://www.science.org/content/article/potential-fabrication-research-images-threatens-key-theory-alzheimers-disease
- ‘Manipulated’ Alzheimer’s data may have misled research for 16 years (telegraph.co.uk)
- Alzheimer’s Disease, and Breast and Prostate Cancer Research: Translational Failures and the Importance to Monitor Outputs and Impact of Funded Research – PubMed (nih.gov)
- Successful therapies for Alzheimer’s disease: why so many in animal models and none in humans? – PubMed (nih.gov)
- Is it Time for Reviewer 3 to Request Human Organ Chip Experiments Instead of Animal Validation Studies? – Ingber – 2020 – Advanced Science – Wiley Online Library
- Seeking alternative animal models for Alzheimer’s disease | National Institute on Aging (nih.gov)
- White Papers – Human Relevant Science
- A human induced pluripotent stem cell‐derived cortical neuron human‐on‐a chip system to study Aβ42 and tau‐induced pathophysiological effects on long‐term potentiation – Caneus – 2020 – Alzheimer’s & Dementia: Translational Research & Clinical Interventions – Wiley Online Library
- Dura, Adelaide; Gribaldo, Laura; Deceuninck, Pierre (2021): EURL ECVAM Review of non-animal models in biomedical research – Neurodegenerative Diseases. European Commission, Joint Research Centre (JRC) [Dataset] PID: http://data.europa.eu/89h/a8fd26ef-b113-47ab-92ba-fd2be449c7eb
- Advancing Alzheimer’s research: A review of big data promises – ScienceDirect
- https://www.j-alz.com/editors-blog/posts/science-based-falsifiability-test-amyloid-hypothesis-ahyp
- HESPEROS HUMAN-ON-A-CHIP® SYSTEM MODELS PRECLINICAL STAGES OF ALZHEIMER’S (hesperosinc.com).
- Who is most at risk of dementia? – Alzheimer’s Research UK (alzheimersresearchuk.org)
- Leroy Hood: The key to treating Alzheimer’s disease may not be a drug – Twin Cities
- https://aspe.hhs.gov/reports/national-plan-2021-update
- https://www.j-alz.com/editors-blog/posts/what-wrong-alzheimers-disease-clinical-research
 For our first blog of 2024, we summarise five key messages from Safer Medicines Trust’s landmark new book, Rat Trap by Dr. Pandora Pound which we are delighted has been shortlisted for a 2024 Lush Prize in ‘Public Awareness’.
For our first blog of 2024, we summarise five key messages from Safer Medicines Trust’s landmark new book, Rat Trap by Dr. Pandora Pound which we are delighted has been shortlisted for a 2024 Lush Prize in ‘Public Awareness’.